サルコイドーシス (sarcoidosis) とは、非乾酪性の類上皮細胞肉芽腫が臓器に認められる疾患である。日本では厚生労働省が認定する特定疾患の1つである。
概念
サルコイドーシスは、リンパ節、肺、皮膚、眼を侵す場合が比較的多いものの[1]、心臓、骨格筋、肝臓、脾臓、唾液腺などが侵される場合もあるように、全身諸臓器に乾酪壊死を認めない類上皮細胞による肉芽腫の結節が形成される、全身性の肉芽腫性疾患である。典型的には若年女性に好発し、肺門部リンパ節腫脹および肺野病変、皮膚、関節および眼症状にて初発する場合が多く、約90%が肺病変を形成すると言われている。
サルコイドーシスには、Th1が関与した過敏性の免疫反応が、背景に存在していると考えられている。サルコイドーシスの免疫病態は、活性化したマクロファージとT細胞の集積であり、その結果が、肉芽腫の形成である。マクロファージが放出するTNF-αは、肉芽腫の形成において重要な役割を担っていると考えられている。また、サルコイドーシスでは、マクロファージ活性化作用を有する、CD4陽性T細胞が放出するIFN-γも、重要な役割を発揮していると考えられている。
しかしながら、1869年にサルコイドーシスの皮膚病変が、イギリスの内科医のジョナサン・ユッチンソン(英語版)らによって報告されて以来、その原因は、なお不明である。
本症の病因として疑われている要因として、サルコイドーシスを引き起こし易い素因を有する宿主が、環境中の何らかの抗原物質(起因体)に暴露された結果として誘導される、Th1タイプの過敏性免疫反応に起因すると考えられている。その起因体の候補としては、例えば、グラム陽性の嫌気性細菌であるアクネ桿菌(英語版)だという説も有る。このような常在菌による感染を起因体だと考える根拠の1つとして、抗菌薬の1種であるミノサイクリンの長期投与によって、皮膚サルコイドーシスが寛解したという報告が有る事が挙げられる。しかし、それこそ、どこにでも見られるような、種々の環境刺激に対して、サルコイドーシスの際で見られるような免疫反応が起きたとする報告も有る。さらには、ストレス等が遠因だとも言われるものの、その真偽は不明である。
疫学
日本での有病率は人口10万人当たり7.5~9.3で、罹患率は平均0.7である。女性に好発する傾向が見られる。アメリカ合衆国での人口10万人当たりの有病率は、男性5.9、女性で6.3である。好発年齢は40歳以下の成人に好発し、20歳代から30歳代に発症のピークが有る。ただし、スカンジナビア諸国と日本では、発症のピークが2峰性を示し、20歳代から30歳代だけでなく、50歳代から60歳代の女性が第2の発症のピークとして見られる。
また、地域別に見ると、北欧や北米、日本では北海道や東北地方など、寒冷地に多く見られる[2]。
病態
サルコイドーシスの肉芽腫形成や進展機序にはTh1細胞による免疫応答の関与が考えられている[3]。有機粒子や無機抗原、病原体などの抗原がマクロファージなどの抗原提示細胞で処理され、MHC class2分子を介してT細胞に提示され、CD4陽性のTh1細胞の活性化と増殖が起こる。T細胞やマクロファージからIL-2やIFN-γ、TNF-α、IL-12、IL-18などのサイトカインやケモカインが産出され肉芽腫形成に関与する。肉芽腫は成熟とともにTh2細胞を介して消退し、TGF-βなどにより線維化をきたす[4][5][3]。さらにTh17細胞がサルコイドーシスの肉芽腫形成や線維化の進展に寄与することが示唆されている[6]。
病理
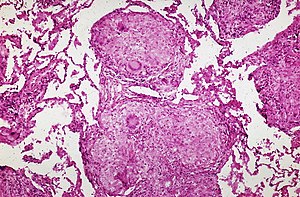 サルコイドーシスによる非乾酪性の肉芽腫病変。ラングハンス型巨細胞を認める。
サルコイドーシスによる非乾酪性の肉芽腫病変。ラングハンス型巨細胞を認める。
サルコイドーシスの病理は多彩だが、リンパ組織や肺に多い肉芽腫性病変、全身性の微小な血管炎(マイクロアンギオパチー)が多いとされている。肉芽腫性病変が肺で発生した場合は、リンパ管に沿うように間質に分布する場合が多いものの、その癒合性、局在部位、臓器特異性によって様々な形態像を見せる。
なお、非乾酪性肉芽腫を形成する異物型巨細胞の細胞質には、星状(英: asteroid)小体やシャウマン(英: Schaumann)小体が見られる場合があるものの、これは本症に特異的ではなく、例えば、結核やベリリウム症などでも認められる。肉芽腫性の病変の大部分は自然退縮するが、硝子化として残存したり、少数例では繊維化へと進展する。ミクロアンギオパチーは肉芽腫が血管壁を侵襲し、血管壁の構造破壊によって発生すると考えられている。病理学的な検討によると、血管壁での肉芽腫の分布は分節的であり、外膜から中膜にかけての分布が多いとされている。
- 肉芽腫性血管炎
- 肉芽腫による血管壁の構成成分の破壊が見られるという病理像を以って定義される。本症では肺、眼、脳、神経などで認められる。
- 病理解剖を行った肺では、弾性型肺動脈から小葉間静脈まで様々な血管で、肉芽腫が認められる。ただし、病理学的には静脈侵襲が目立つ傾向にある。血管での肉芽腫の分布は分節的で、血管外膜から中膜にかけて多く分布する。巨細胞は星状(asteroid)小体やシャウマン(Schaumann)小体など、細胞内封入体を有する場合がある。肉芽腫の中心部は、主にCD4陽性のリンパ球で構成され、辺縁部はCD8陽性細胞で構成される。なお、好中球浸潤やフィブリノイド壊死は認められない。また、ミクロアンギオパチーが共存している場合もある。
- ミクロアンギオパチー
- 当初のミクロアンギオパチーは、眼底における細動脈の狭小化、白鞘化、静脈周囲炎、細動脈拡張などの変化に付けられた名称であった。その後、全身の微小血管変化に用いられるようになった経緯を有する。
- ミクロアンギオパチーには、類上皮細胞やマクロファージから分泌される内皮細胞増殖因子の関与も疑われる。病理学的には電子顕微鏡像で骨格筋、網膜血管、気管支粘膜、心筋、あるいは皮膚等における、毛細血管や細静脈に内皮細胞の核の濃縮、変性、基底膜の多層化を特徴とする。
- 肉芽腫の転帰
- サルコイドーシスの肉芽腫は多くの場合、自然消退するものの、中には硝子化、線維化へと進展する物が有る。消退の過程においては類上皮細胞が消失し、巨細胞が線維化の中に残る場合が多い。
脳サルコイドーシス
脳サルコイドーシスの病理所見は硬膜や軟髄膜の肥厚、視床下部、下垂体茎あるいは脳神経周囲にも病変が認められる。非乾酪性肉芽腫性病変であり、肉芽腫性炎症が髄軟膜、脳室、近接する脳実質、脊髄などに認められる。非乾酪性であるが、中心部に線維化を認めることがあり、限局性に壊死を認めることもある。また小血管にも肉芽腫が認められる。通常は類上皮細胞と多核巨細胞が認められる。多核巨細胞はCD68陽性で、細胞体の周囲に核が偏在するラングハンス型の場合もある。サルコイドーシスに特徴的とされるアステロイド小体またはシャウマン小体は必ずしも特異的ではない。真菌や結核が認められないことを組織学的あるいは培養で確認することも重要である。神経系以外のサルコイドーシスが確定していても中枢神経系に同一の病変があるとは限らない[7]。
クリプトコッカス髄膜炎や結核性髄膜炎など他疾患に注意が必要である。
脊髄サルコイドーシス
脊髄サルコイドーシスでは脊髄は急性、亜急性期には浮腫により腫大するが、慢性期には脳実質の破壊によって萎縮を示すようになる。病変はくも膜と脊髄実質内、神経根に認められる。これらの部位は活動期にはリンパ球・マクロファージを主体とする炎症細胞浸潤が強く、髄膜・脊髄・神経根炎の所見を示す。病変は髄膜から血管周囲腔に沿って脊髄実質内に広がっていく傾向がある。髄膜および脊髄実質内に巨細胞を伴う非乾酪性上皮肉芽腫病変を認め、炎症の陳旧化した場所では硝子化、線維化した小結節もみられる。ときに血管病変からの二次的循環障害によると思われる脊髄実質の壊死も認められる。また髄外に肉芽腫を形成することもある[8][9]。
末梢神経サルコイドーシス
サルコイドニューロパチーでは神経上膜を中心に肉芽腫と壊死性血管炎の所見に加え、血管炎ニューロパチーで見られるような、急性軸索変性像と神経束ごとの有髄神経線維密度の偏りが報告されている[10]。
筋サルコイドーシス
サルコイドミオパチーにおける類上皮細胞肉芽腫は他臓器と同様で、中心部に活性化されたCD4陽性T細胞、類上皮細胞やマクロファージなどのCD68陽性細胞、ラングハンス巨細胞が多数集簇して存在し、周辺部にCD8陽性T細胞やB細胞が見られるのが特徴である[4][11][12]。類上皮細胞はリンパ球に比べるとやや扁平で大きく、核はクロマチンに乏しい。CD68陽性細胞はカルパインやカテプシンB、ユビキチン・プロテアソームなどのタンパク分解酵素を強く発現する。これは全身の肉芽腫性病変に共通した所見である[11]。肉芽腫は筋周膜や筋内鞘の小血管に形成され、周囲の筋線維を破壊しながら進展して形成していく。増大した肉芽腫では、周辺部から中心部に向けて線維化が進展し、硝子化病変となる。そして高度の線維化を残して自然消滅する。この肉芽腫の形成、消退、線維化のサイクルはearly、premature、mature、healingの各ステージに分けられ、症例や病変部位により種々のステージの肉芽腫が観察される[11][12]。
またサルコイドミオパチーでは臨床病型に筋病理学の相違点が知られている。腫瘤型では筋周膜や筋内鞘などの間質の血管周囲を中心に肉芽腫が形成され、様々なステージの肉芽腫が認められる。筋線維内に炎症細胞が浸潤し、また筋束内の肉芽腫の増大とともに肉芽腫に接する筋線維が圧排されることで、筋線維の崩壊にいたる。病変の一部で筋鞘膜蛋白により周囲を囲まれた肉芽腫がみられることがあり、肉芽腫が筋線維内に形成されていることが示唆される[11]。また血管壁に浸潤して肉芽腫性血管炎を認めることもある。病変部位から離れた筋束の筋線維は正常である。
一方、ミオパチー型では、限局性の肉芽腫病変が多巣性にびまん性に形成され散在するが、時に見られないこともある。筋線維の著明な消失から筋束の基本構築の崩壊が認められる。残存する筋線維は高度の大小不同を認め、筋線維の壊死や再生、小角化線維、小円形線維が認められる。肉芽腫性病変から離れた筋束の筋線維でも変化が認められる。その他、縁取り空胞や赤色ぼろ線維、分葉線維、cytoplasmic bodyなどを有する線維が散見される[13][11]。また筋周膜や筋内鞘の高度線維化と脂肪置換を認め、血管周囲の炎症細胞浸潤や肉芽腫性血管炎がみられる。
腫瘤型とミオパチー型は筋病理像が大きく異なる。腫瘤型がミオパチー型に進展する症例は殆どなく、腫瘤型では腫瘤が全身性に形成されても筋力低下がみられない。一方、ミオパチー型では四肢対称性に筋力低下を認めることから、ミオパチー型の高度のびまん性筋崩壊には腫瘤型と異なり、種々の自己免疫機序や内分泌因子、液性因子などの二次的な機序の関与も示唆される[13][11]。実際、ミオパチー型においてTh2免疫応答を基盤としたM2分化型マクロファージが慢性型に関与することが報告されている[14]。
自然経過
サルコイドーシスの臨床所見、自然経過、予後は極めて多彩である。サルコイドーシスの症例の多く(28~70%)は、自然治癒するとされている。その場合は、2年以内に病変が消失する。予後不良因子としてはlupus pernio、慢性ブドウ膜炎、40歳以降での発症、慢性高カルシウム血症、腎臓の石灰化病変、黒色人種、進行性肺サルコイドーシス、鼻粘膜病変、嚢胞性骨病変、神経サルコイドーシス、心筋病変、慢性呼吸不全とされている。1~5%が本症のサルコイドーシスの病変で死亡する。典型的な死因は、進行性の呼吸不全、中枢神経系や心臓病変による。日本においてはサルコイドーシスによる死亡の77%が心筋病変によるのに対して、アメリカ合衆国では心筋病変よりも肺病変による死亡の方が多い。
症状
- 心サルコイドーシスによって発生した不整脈は、致死的な場合がある。
- 肺での両側肺門リンパ節腫脹 (英語: bilateral hilar lymphadenopathy;BHL) は、サルコイドーシスで特徴的とされる。咳を症状として訴える。
- 眼症状として、ブドウ膜炎を合併する場合がある。目のかすみ症状を訴え、飛蚊症、視力低下・眼圧上昇を来す事がある。
- 皮膚症状として、結節性紅斑などを認める場合がある。
検査
サルコイドーシスの診断で重要な点として、サルコイドーシスの組織学的確認をする事、臓器病変の広がりとその程度を評価する事、病態が安定しているか進行性かを評価する事、治療が患者に利益を与えるかを評価する事の4点が挙げられる。なお、サルコイドーシスの診断のために、以下のような検査を実施する場合がある。
胸部X線
サルコイドーシスの胸郭内病変は肺門、縦隔のリンパ節病変と肺野病変に大別できる。胸部X線の所見に基づき病期分類があり予後と関連する。両側肺門リンパ節腫脹(BHL)が診断を強く示唆する特徴的な所見である。健康診断などで胸部X線撮影を行った結果、偶然発見される場合もある。BHL以外の胸部X線検査所見としては上肺野優位の粒状影や斑状影を中心としたびまん性肺野陰影、中枢部気管支血管周囲束の不整肥厚、進行期にみられる上肺野優位の収縮を伴う線維化病変がある。胸部X線検査でサルコイドーシスのリンパ節石灰化を認識できることは稀である。胸部X線撮影で観察された両側肺門リンパ節腫脹(BHL)と、肺野病変の有無によって5つのstageが存在する。ACCESSでは各病気の頻度としては、I期39.7%、II期36.7%、III期9.8%、IV期5.4%と報告されている。ステロイド系抗炎症薬の投与を行えば、短期的には病変の消失や縮小には寄与するものの、長期的な有効性は明らかになっていない。II期、III期において自覚症状、呼吸症状が認められる場合に、ステロイド系抗炎症薬の使用が検討される。
| 胸部X線像病期 |
内容
|
| stage 0 |
正常な胸部X線像
|
| stage I |
両側肺門リンパ節腫大
|
| stage II |
両側肺門リンパ節腫大+肺陰影
|
| stage III |
肺陰影のみ(両側肺門リンパ節腫大なし)
|
| stage IV |
肺線維
|
胸部X線写真の所見で、サルコイドーシスの予後を、ある程度予測できる。stage Iの場合は胸部X線写真所見は通常、自然に改善ないし安定化する。肺門リンパ節腫大の持続は、活動性病変が続いている事を意味するわけではないため、経過観察が行われる。自然寛解は16~39%の症例で発症後6~12ヶ月の間で認められている。自然寛解の85%以上は、発症後2年以内に起こる。自然寛解した症例ないし、安定化した症例で、後に再燃が認められた症例は、わずかに2~8%程度である。2年以内に自然消退しない場合は、慢性ないし持続性の経過をとる可能性が高まる。よって肺サルコイドーシスの長期観察は、発症後2年間に最も集中的に行われるべきである。stage Iでは6ヶ月毎、stage II~IVでは3~6ヶ月とより頻回の評価が必要である。治療を行った場合は、治療後最低3年間は追跡をする。また持続性のstage II~IVでは、少なくとも数年ごとに無期限に追跡をするべきとされている。重症の肺外病変がある場合も、長期の追跡が必要である。
胸部CT
サルコイドーシスの胸部CT所見としては小粒状影・気管支血管周囲間質の肥厚像、リンパ路に沿った粒状影が特徴的とされている。時に見られる所見として結節影、塊状影、均等影が知られている。非典型的な所見としては胸水、定型的な蜂巣肺を伴う線維化病変がある。ここではリンパ節病変、肺野病変、上腹部の異常所見に分類して述べる。サルコイドーシスの90%で胸部画像で異常所見が認められる[15]。神経サルコイドーシスに限っても60~70%で胸部画像に異常が認められる[16]。
リンパ節病変
BHLが特徴的なリンパ節病変である。サルコイドーシスは両側性が原則であり、片側性の場合は肺がんを疑う。サルコイドーシスのリンパ節も慢性期は石灰化を伴うことが少なくない。しかし結核感染後などに見られる均一で非常に高吸収を呈するリンパ節は稀でありある。
肺野病変
サルコイドーシスの肺野病変は病期や罹患期間により違いがあるが基本的には肉芽腫による微細粒状影と線維化により構成される。粒状陰影は多くが3~4mmくらいまでの微細な病変でリンパ路に沿って分布する傾向が強い。肺内でリンパ路は臓側胸膜直下、小葉間隔壁内、肺静脈周囲、気管支・肺動脈周囲の間質に分布しており、粒状影の好発部位もこれらの構造に一致する。肺胞隔壁にはリンパ管は存在せず、肺胞領域の粒状影は比較的少ないのが特徴である。微細粒状影は粟粒結核や甲状腺癌に血行性転移などでも認められるため鑑別にはリンパ路との関係が重要である。
サルコイドーシスの肉芽腫性病変はここの結節がある程度大きく病変の密度が低いときは粒状影を呈するが個々の結節がCTの分解能よりも小さい場合や病変が密集する場合は肺野濃度が上昇を呈しうる。肉芽腫に付随するマクロファージの浸潤の程度によりすりガラス状陰影から均等影(コンソリデーション)まで幅広い吸収値を呈しえる。広範な濃度上昇の場合は、間質性肺炎や肺水腫などが鑑別にあがる。濃度上昇した部位に比較的大きな粒状影が認められる場合が多い。斑状や限局性の濃度上昇では腫瘍性病変が鑑別にあがる。限局性病変の周囲には微細粒状影が認められることが多い。
サルコイドーシスの肉芽腫性病変は痕跡を残さずに消失することが多いが線維化が残存することがある。このため、慢性例では容積減少、牽引性気管支拡張など間質性肺炎類似の所見が生じうる。
上腹部の異常所見
上腹部の異常所見ではリンパ節腫脹が多く、膵臓周囲や胃噴門周囲で出現率が高い。
呼吸機能検査
肺野病変を伴わない場合は呼吸機能に異常は認めない。肺野病変を合併すると何らかの呼吸障害が認められる。線維化病変が進行すれば間質性肺炎と同様に拘束性換気障害が認められる。気道壁やその周囲に肉芽腫が出現すれば閉塞性換気障害を示す。
心電図
心サルコイドーシスは房室ブロックや心室頻拍などの重症不整脈、重症心不全の原因となる。心サルコイドーシスの心電図では軸変異、ST-T異常、異常Q波、刺激伝導障害、上室性および心室性不整脈など様々な所見が認められる。特に完全右脚ブロックや房室ブロックを呈する例が多い。
心臓超音波検査
心サルコイドーシスの心臓超音波所見は多彩である[17]。サルコイドーシスによる肉芽腫性炎症の活動期には浮腫を伴うリンパ球浸潤や類上皮細胞肉芽腫によって、病変部の心室壁肥厚と壁運動低下を生じる。肉芽腫性炎症が線維化へ移行すると、病変部の心室壁菲薄化とエコー輝度の上昇が認められるようになり、時に心室瘤形成がみられることもある。病変が局在する場合、局所的な壁運動異常や心室瘤として観察される。左室全体にびまん性に波及した場合には拡張型心筋症様の所見を呈するようになる。また、時に肥大型心筋症と同様に非対称性中隔肥厚を示す症例も存在する。
心サルコイドーシスでは早期例では心サルコイドーシスのリモデリング予防効果、中等度では心機能改善効果がステロイドにはあるのかもしれない[18]。
ACE
ACEはアンギオテンシンⅠやブラジキニンのような種々のペプチドを切断する。ヒトではACEは血管内皮に膜結合型として広く分布し、健常者では内皮型ACEが細胞表面から切り取られて血中に可溶型ACEが循環している。サルコイドーシスでは肉芽腫内の類上皮細胞を含む単球系細胞からACEが産出される。血清ACE値は疾患活動性マーカーであり肉芽腫量を反映している[19][20][21]治療によって低下が認められる。酵素活性で測定しているためACE阻害薬内服時は低値になる。
リゾチーム
マクロファージで産出がさかんでありサルコイドーシスの疾患活動性マーカーである[22][23]。
sIL-2R
悪性リンパ腫など造血腫瘍のほか、乳がんなど非造血腫瘍、結核などの感染症、膠原病で高値を示す。サルコイドーシスでも疾患活動性マーカーでありT細胞活性化の指標と考えられている[24][25]。サルコイドーシスの診断基準2015では、血清可溶性インターロイキン2レセプター(sIL-2R)高値も、特徴的な検査所見として記載されている。
ツベルクリン反応
サルコイドーシスによる細胞性免疫低下のために、ツベルクリン反応が陰転化する事が知られているものの、サルコイドーシス診断基準2015では参考所見から除外された。
MRI
脳MRI
- 髄膜病変
軟膜病変の好発部位は脳底部、後頭蓋窩、視交叉であり、同部位にびまん性または結節性のGd増強効果を認める。軟膜病変がGd造影剤を使用しないと認識できないことが多い。Gd造影剤はT1短縮効果を持つため、造影後の画像評価は一般的にはT1WIで行われるが、造影後のFLAIR画像は軟膜の異常造影効果をT1WIより明確に描出することができる。またGd-T1WIでは脳表の静脈が高信号を示すがFLAIR画像ではこの信号が抑制されるため髄膜炎所見がよりわかりやすい[26]。病変は豊富な肉芽腫を反映しT2WIでは低信号になることが多いとされている。また髄膜病変は拡散制限は乏しく、悪性リンパ腫や髄膜腫との鑑別に有用である。硬膜に腫瘤を形成することもある。硬膜の腫瘤は豊富な線維成分を反映し、T2WIでは低信号を呈し、比較的均一なGd増強効果を認める[27][28]。
- 下垂体病変
下垂体と視床下部は脳底部に位置しており神経サルコイドーシスの好発部位の一つである。下垂体柄の肥厚とGd増強効果や下垂体後葉T1WI高信号の消失などが知られる。下垂体後葉T1WI高信号の消失は尿崩症に対応する。
- 水頭症
サルコイドーシスはしばしば水頭症をきたすことがある。交通性水頭症と非交通性水頭の両者ともありえる。SSFP(3D-stedy-state free precession)を撮影することで狭窄部位が同定できることがある。交通性水頭症は軟膜病変により脳脊髄液の吸収が阻害されることで生じる。
- 血管病変
サルコイドーシスの血管病変は稀とされているが剖検例ではしばしば肉芽腫の血管浸潤が認められる[29][30][31][32][33]。病理学的にはサルコイド肉芽腫は軟膜から穿通動脈のVirchow-Robin腔に沿って脳実質に浸潤することが知られている。血管周囲腔の肉芽腫は好中球などの炎症細胞とともに血管壁に浸潤しフィブリノイド壊死を起こす。これにより血管壁の内弾性板が破壊され血管の閉塞や破綻が生じ脳梗塞に至る[31][34][35][36]。また心サルコイドーシスも心筋症の結果心原性脳塞栓症を起こす。
- 脳神経病変
しばしばGd増強効果が認められる[37][38][39]。
脊髄MRI
脊髄サルコイドーシスのMRI所見はJungerらによって4期に分類される[40]。第1期は脊髄腫大はないがGd造影で脊髄表面の軟膜が線状に造影される。これはくも膜下腔の炎症の表現である。第2期は髄膜から血管周囲腔を通って脊髄実質内に炎症が広がる。この時期には脊髄が腫大し、Gd造影では造影されない場合からびまん性に造影される場合まで様々である。第3期では脊髄が腫大する場合と正常に復する場合があり、Gd造影では髄内に単発あるいは多発する結節性の造影が認められる。第4期は慢性期で脊髄組織の壊死のため脊髄は萎縮してGdでは造影もされなくなる。すべての脊髄サルコイドーシスが上記のような経過をとるとは限らず、初期から腫瘤を形成する場合もある。なお軟膜のGd増強効果は背側面から髄内に浸潤性に進展することが多いが、特に三日月状に広がった軟膜のGd増強効果と中心管のGd増強効果が合わさってみられるtrident signは脊髄サルコイドーシスの89%で認められる[41]。脊髄サルコイドーシスは頸部脊柱管狭窄の強い部位に後発する。
筋MRI
腫瘤型の筋サルコイドーシスではT1WI、T2WIの横断像で腫瘤の周辺部分の肉芽腫病変が高信号、線維化している中心部が低信号を示す。Gd造影では周辺部が著明に増強され、星型の中心部は造影されずdark starと言われる[42]。冠状断では、中心部の低信号層が両側の高信号層に挟まれた三層構造がみられる。三層構造をthree stripesといい、腫瘤型の筋サルコイドーシスの疾患特異度が高い所見である[43]。あたかもthree stripesにみえる所見はしばしばサルコイドーシス以外の筋炎でも認められることがある。特に低信号病変認められない時は注意が必要である。治療後はGd増強効果の消失と腫瘤性病変の縮小・消失が認められる。ミオパチー型は、筋萎縮所見とともにT1WIでは低信号、T2WIで高信号の多発結節状の非特異的病変を認め、しばしば淡いGd増強効果を示す[44][43]。
シンチグラフィー
- ガリウムシンチグラフィー
放射性同位体である67Ga(英語版)のクエン酸塩を注射して、体外で67Gaから放出される放射線を検知して画像化する事で、炎症が起きている箇所を検索する検査である。この検査によって得られた、縦隔や両側肺門上部に、67Gaの集積が亢進した結果の画像であるラムダサインや両側耳下腺、両側涙腺の左右対称性の集積亢進の結果の画像であるパンダサインはサルコイドーシス診断を支持する[45][46][47]。
PET
18F-FDG/PETでは病変に一致して集積増加を認めることが多いため、サルコイド病変部位の同定に有効な可能性がある。サルコイドーシスで観察されるFDG集積像は病変部における活性化マクロファージなどの炎症細胞浸潤を反映したものと考えられる[48]。炎症細胞への18F-FDG集積機序はがん細胞とほぼ共通であり細胞膜のグルコーストランスポーターのうち、GLUT1やGLUT3の増加、ヘキソキナーゼの活性化によるとされている。筋サルコイドーシスでは全身の筋肉に結節が多発する傾向があり18F-FDG/PETでの所見はleopard-man[49]、tiger-man[50]と呼ばれることがある。サルコイドーシスの脊髄病変にも集積効果があり有用という報告もある[51]。しかし頚椎症でも陽性を示すことがあり疾患特異性は低いと考えられる[52]。
気管支鏡
- 気管支鏡観察
気管支鏡観察所見では粘膜発赤、網目状血管増生、黄白色小結節などが知られている。網目状血管増生はミクロアンギオパチーの所見という解釈もある。
- 気管支粘膜生検(EBB)
TBLBにEBBを追加すると診断率が向上するという研究がある[53][54]。
- 経気管支的肺生検 (TBLB)
TBLBで4ないし5個の肺生検を行えば、診断率は40~90%である。
- 気管支肺胞洗浄(BAL)
殆ど侵襲なしに終末細気管支、肺胞領域からの細胞、吸入粉塵、病原物質、液体成分を採取できる。サルコイドーシスのような、びまん性肺疾患の場合、BALは中葉から行われる。サルコイドーシスにおいては多くの場合、気管支肺胞洗浄液(BALF)中の総細胞数、リンパ球比率、CD4/CD8比が増加する。総細胞数やリンパ球比率の上昇は、種々のびまん性肺疾患で観察されるものの、CD4/CD8比の上昇が加わった場合はサルコイドーシスを疑う大きな根拠とされる。BALF中のCD4/CD8比が3.5以上に上昇すれば、サルコイドーシス診断の感度は52%、特異度は94%であり、CD4/CD8比が5.0では特異度97%となり生検しなくともサルコイドーシスと診断できるという報告がある[55]。CD4/CD8比が4.0以上ならば感度59%、特異度96%という報告もある[56]。そのため生検を実施していない症例では、診断の補助となると考えられている。ただし、感度は低いためBALFが正常であっても、サルコイドーシスは否定できない。眼サルコイドーシス所見、BHL、血清ACE高値といったサルコイドーシスに典型的で特徴的な臨床症状・所見を満たしかつBALでCD4/CD8比が3.5以上ならば組織診断群に匹敵する診断的価値があるという意見もある[57]。なお、BALFは喫煙の影響を受け、喫煙によって総細胞数は3~4倍に増加し、マクロファージの比率が増加し、リンパ球比率が低下する。CD4/CD8比も低下するとされている。悪性リンパ腫はCD4/CD8比が低値であることが多いが高値を示すこともあり病型によって異なる可能性がある[58]。すなわちサルコイドーシスと悪性リンパ腫の鑑別はBALのCD4/CD8比のみでは十分ではない可能性がある。
神経サルコイドーシス全体ではBALのリンパ球増多やCD4/CD8比の上昇が81.2%の症例で認められる[59]。脊髄サルコイドーシスでは83.3%とほぼ同様であった[60]。
脳脊髄液検査
神経サルコイドーシスと他疾患の鑑別のために脳脊髄液検査は重要である。造影MRIでびまん性増強効果を呈する神経サルコイドーシス症例の脳脊髄液検査の結果が報告されている[61]。単核球優位の細胞数増加、蛋白増加、糖低下が認められる。糖が低下する原因としてはリンパ球やマクロファージによる糖の消費という考え方がある[62][63]。脳脊髄液糖の高度低下は細菌性髄膜炎などとの鑑別を難しくすることや、67%の症例で40mg/dl以下の脳脊髄液糖の低下が認められたという報告もある。
脳脊髄液のACEが増加することはしばしば報告されている[64][65]。しかし感度66.7%、特異度67.3%であり有用とは言い難い[66]。脳脊髄液のリゾチーム、β2ミクログロブリン、IgG index、sIL-2Rなどの高値やオリゴクローナルバンドの存在も報告されている[67][68]。悪性リンパ腫は髄液IL-10が高値を示すため鑑別に有効な可能性があるが[69]、その他の炎症性疾患でも高値になりえる[70][71]。
治療
サルコイドーシスの臨床所見、自然経過、予後は極めて多様である。サルコイドーシス全体では60%以上に近い症例で自然寛解が得られる。しかし、30%程度の症例で慢性、ないし進行性の経過をとる。サルコイドーシスによって死亡する患者は、サルコイドーシス罹患者の5%以下であり、その死因は進行性の呼吸不全、中枢神経病変や心臓病変である。心臓や中枢神経に病変が及んだ例や、肺線維症を起こしてしまった場合は予後が悪い。
「ATS/ERS/WASOGによるサルコイドーシスに関するステートメント」によると、心臓病変、中枢神経病変、治療抵抗性の眼病変、高カルシウム血症を認めた場合は、積極的な治療適応が有るとしている。治療法は、ステロイド系抗炎症薬を経口投与する方法が一般的である。なお、びまん性浸潤型皮膚サルコイドーシス(Lupus pernio)[72] や、神経サルコイドーシスには、抗TNF-α抗体であるインフリキシマブの注射投与が有用だと報告されている[73]。なお、皮膚病変についてはサリドマイドも注目されている。
サルコイドーシスで見られる臓器の障害
肺サルコイドーシス
サルコイドーシスは経過中に90~95%に肺実質病変を伴うことが知られている。日本での本症の特徴としては、50~70%が胸郭内病変で発見されている。無症候性の両側肺門リンパ節腫脹(BHL)などで、健康診断で指摘される場合が多い。進行例では、乾性咳嗽、労作時呼吸困難などが認められ、肺の線維化が認められる場合もある。本症の約2/3の症例に自然寛解認められるのに対して、10~30%の症例では慢性または進行性に経過する。発症年齢が40歳以上、肺外病変の存在、lupus prernioを予後不良因子とする報告も存在する。これに対して、肺サルコイドーシスの活動性指標は、予後因子ではない事が知られている。病理学的にはサルコイドーシスの類上皮細胞肉芽腫は、通常、気管支・血管束、小葉間隔壁、胸膜下リンパ流路に沿って分布する。肉芽腫の分布は両側性で上葉に著しい。基本的には0.2 mm程度の大きさの肉芽腫であり、これらが融合し、塊状陰影や線維化を形成すると考えられている。画像上は間質病変のパターンをとり、進行例では上葉に肺線維症の初見を示す場合がある。肺のみにならず、多臓器に発達した肉芽腫は70%以上自然退縮するが、一部の進行例は線維化と蜂窩肺形成する。そのためKL-6は進行例の活動性マーカーとされている。
病理学的には発症1年以内の症例で、画像所見の有無に関係無く95%で肺に肉芽腫が認められ、その率は、より経過が長い例では50%以下である。そのためサルコイドーシス確定診断のために、肺病変の検出が試みられる場合がある。経気管支肺生検(TBLB)、気管支粘膜生検(EBB)、縦隔リンパ節の経気管支吸引肺生検(TBNA)、気管支肺胞洗浄(BAL)などが用いられる。気管支拡張症を伴い、アスペルギローマを合併する場合は、イトラコナゾールのような抗真菌薬が使用される場合がある。
眼サルコイドーシス
サルコイドーシスの約50%に眼病変が生ずるとされており、その多くはぶどう膜炎と呼ばれる眼内炎症疾患である。ぶどう膜炎の原因疾患は、かつてはベーチェット病が最も多かったものの、近年はサルコイドーシスが最も多い。眼サルコイドーシスでは非特異的な眼内炎症病変の他に、特徴的な眼病変が混在している。サルコイドーシスの診断基準では、前部ぶどう膜炎、隅角結節、周辺部虹彩前癒着、硝子体混濁、網膜周囲血管炎、網脈絡膜滲出斑および結節、網脈絡膜広範囲萎縮病変などが特徴的な所見とされている。サルコイドーシスでは長期にわたる慢性の眼内炎症によって、白内障、緑内障、嚢胞様黄斑浮腫、黄斑上膜などの視力の低下につながる重篤な合併症を生ずる場合がある。白内障は、サルコイドーシスの約半数に認められる。緑内障は、約20~30%に認められる。眼圧の上昇の原因としては、隅角結節による房水流出障害、テント上PASによる房水流出障害、ステロイド系抗炎症薬の副作用などが挙げられる。なお、隅角結節による眼圧の上昇には、ステロイド系抗炎症薬が有効であるものの、その副作用の有無について注意を払う必要がある。
- 前部ぶどう膜炎
- 前部ぶどう膜炎とは、虹彩あるいは毛様体などの前眼部に発生した炎症を言う。前房水中に炎症細胞浸潤が認められれば、前部ぶどう膜炎と診断される。
- ただし、これはサルコイドーシスに限らず、ベーチェット病、vogt-小柳-原田病など、殆ど全てのぶどう膜炎で見られる非特異的な眼病変である。
- 肉芽腫性前部ぶどう膜炎
- 豚脂様角膜後面沈着物や虹彩結節が認められる前部ぶどう膜炎を肉芽腫性前部ぶどう膜と言い、サルコイドーシスに特異的と考えられている。ベーチェット病など非肉芽腫性血管炎では、角膜後面沈着物は炎症細胞びまん性に沈着する。これに対して、サルコイドーシスの場合は炎症細胞が集簇し、大型の角膜後面沈着物を形成する。これを豚脂様角膜後面沈着物と言う。
- 虹彩結節
- 虹彩結節は虹彩に生じる肉芽腫であり、瞳孔縁のKoeppe結節や虹彩実質のBusacca結節が知られ、特異度の高い所見とされている。
- 隅角結節、テント上虹彩前癒着(テント上PAS)
- 虹彩と角膜が合わさる部分である前房隅角を、隅角鏡で観察する。線維柱帯に、隅角結節(肉芽腫)やテント上虹彩前癒着(肉芽腫の瘢痕)が認められる場合がある。
- 硝子体病変
- ぶどう膜炎では炎症細胞が硝子体に浸潤して、硝子体混濁を起こす。非肉芽腫性ぶどう膜炎ではびまん性の硝子体混濁が起こるのに対して、サルコイドーシスでは硝子体中に類上皮性肉芽腫を形成した結果として数珠状の硝子体混濁が起こる。
- 脈絡膜炎
- ぶどう膜炎では炎症細胞が網膜や脈絡膜に滲出した結果、眼底の白斑あるいは滲出斑と呼ばれる病変が出現する。白斑、滲出斑、出血が黄斑部に及ぶと、高度の視力低下の原因となる。ただし、これはベーチェット病、結核、交感性眼炎、サイトメガロ網膜症などでも認められる非特異的病変である。
- 網膜静脈周囲炎
- ぶどう膜炎の網膜血管炎は、幾つかのパターンが知られている。ヘルペスウイルスによる急性網膜壊死では動脈炎が主体、サルコイドーシスや結核では静脈炎が主体、ベーチェット病では動脈、静脈が同様に広範囲に障害され、vogt-小柳-原田病では網膜血管炎は認められない。サルコイドーシスの静脈周囲炎は、網膜静脈に沿って結節状の白色浸潤が所々に生じる。この竹の節状の網膜静脈周囲炎はサルコイドーシスに特異的と考えられている。
神経サルコイドーシス
サルコイドーシスの中で神経系に病変が認められるものを神経サルコイドーシスという。神経症状は全サルコイドーシスの5%程にのみ認められる、比較的な稀な合併症である。神経サルコイドーシスの約50%は神経症状を初発とするため、診断が難渋する場合が多い。長期観察では、神経サルコイドーシスの90%が神経系以外の臨床症状を示すと報告されている。剖検例では25%程の無症候性サルコイドーシスが認めらる[15]。逆に神経サルコイドーシスの20%程度は全身性の症候を欠く英語: isolated sarcoidosisである。血管壁と軟膜が神経サルコイドーシスの初発と考えられている。病変の進展メカニズムとしては軟膜や血管壁の肉芽腫によって、血液脳関門の破綻が発生し、血管周囲腔(Virchow-Robin腔(英語版))に肉芽腫が侵入し、血管周囲腔に沿って脳実質に進展してゆくと考えられている。血管周囲腔が脳底部で特に大きいため、視床下部、第三脳室、視神経、脳幹から出る脳神経(特に顔面神経)が、障碍され易いと考えられている。その過程や肉芽腫性血管炎によって虚血性変化、脳梗塞も起ると考えられている。中枢神経系では神経サルコイドーシスの肉芽腫性病変は軟髄膜、脳神経、脳実質などに認められる。脳脊髄液循環障害により水頭症をきたすこともある。神経サルコイドーシスは単一の神経障害ではなくいくつかの神経症候を併発することが多い。頻度の多い順に脳神経障害、頭痛、末梢神経障害、運動失調、筋力低下、めまい、認知機能障害、髄膜炎、痙攣、意識障害、水頭症、筋症となる。
脳サルコイドーシス
- 脳神経障害
脳神経は末梢神経であるため末梢神経サルコイドーシスに分類されることもある。顔面神経、視神経が多く、三叉神経、内耳神経が続く。顔面神経麻痺は小肉芽腫が顔面神経の神経外膜や神経周膜に浸潤することで生じ、軟髄膜の肉芽腫が浸潤することで生じることもある。ベル麻痺は再発や両側の発症は稀であるが、サルコイドーシスでは再発や両側性障害が認められる。なお両側性障害は必ずしも同時発症とは限らない。視神経障害では乳頭炎や球後視神経炎を生じ、眼球内の病変を認めることもある。眼窩に限局性の肉芽腫を生じることもあり、眼球突出や複視を認める。特に外眼筋が障害されることもある。MRIにおいては視神経の肥厚や増強効果が認められる。多くは亜急性に進行し視神経炎に類似する症候を呈する。進行性の視神経萎縮を認めることもある。三叉神経障害では感覚障害や三叉神経痛をおこす。内耳神経障害はしばしば両側性難聴を示す。機序は不明であるが血管炎を生じコルチ器が障害されると推定されている。その他海綿静脈洞の障害なども知られている。
- 軟髄膜病変
軟髄膜では肉芽腫あるいはそれに関連した血管炎を認める。臨床的には髄膜刺激徴候を認め、特に緩徐進行性の頭痛を認めるが項部硬直は顕著ではない。全身性サルコイドーシスの治療中の生じた軟髄膜病変は神経サルコイドーシスとは限らないため注意が必要である[7]。結核性髄膜炎、真菌性髄膜炎、癌性髄膜炎などの可能性がある。慢性髄膜炎を原因とする急速進行性認知症の報告もある[74]。慢性髄膜炎は治療可能な認知症であるため、これが疑われた場合は、造影MRIと髄液検査で髄膜炎の診断を行い、早期治療が望まれる。
- 脳実質病変
軟髄膜のサルコイド病変が脳実質へ波及することで単発あるいは多発性の脳実質病変を認める。肉芽腫は脳表から脳実質に入る小血管の周囲腔に沿って脳実質に至ると考えられている。血管自体にも肉芽腫性血管炎や類線維素変異を認めることもあるが多くはない。脳生検などで肉芽腫性血管炎を認めた場合は他疾患との鑑別が難しくなる。頭部MRIでは脳実質病変は3つに分類される[75]。
- 視床下部-下垂体系の障害
下垂体よりも視床下部の肉芽腫性病変の方が多い。
治療
プレドニゾロンを60 (mg/day)で開始し、6か月で20 (mg/day)まで減量し、その後20 (mg/day)で2年間維持するという方法はよくとられるが、これは多くの施設で20 mg前後で再発を起こしているという経験に基づくものである。2年間安定していれば5 mgごと慎重に減量し、全投与を4年程度とするのが一般的である。ステロイド系抗炎症薬による反応が充分でない場合や、ステロイドの副作用により治療継続困難な場合は、シクロフォスファミド、メソトレキセート、アザチオプリン、ミコフェノール酸モフェチルなどが用いられる。しかし神経サルコイドーシスにおいては、免疫抑制剤の使用に関しても比較論文は存在しない。治療効果が予測できないため免疫抑制剤は複数使用してから免疫抑制剤耐性と考えるべきとの意見もある。しかし神経病変の場合は他の臓器よりも不可逆的な変化が短い期間で生じ易く、治療抵抗性、遷延性と判断するタイミングが早い傾向がある。神経学会では3か月から1年以上で遷延性とすることが多いが、日本サルコイドーシス学会では1年から5年以上で遷延性とすることが多い。治療抵抗性の場合は、エンドキサンパルス療法、インフリキシマブ療法、サリドマイド療法が用いられる場合もある。
脊髄サルコイドーシス
脊髄サルコイドーシスは極めて稀であり、サルコイドーシス全体の1%以下と言われている[76][77]。特に病変が脊髄のみ存在するisolated spinal cord sarcoidosisは非常に稀であり[78]、通常は他臓器病変が認められる。臨床症候としては病変レベルに相当した脊髄症候、すなわち運動麻痺(対麻痺、四肢麻痺)、レベルをもつ感覚障害、膀胱直腸障害を呈する。中下位頸髄と中下位胸髄病変が多い。特に中下位頸髄を中心として数椎体以上の広がりを持つものが多い。頸髄病変を呈する場合は上肢よりも下肢の症候が目立つ場合が多い。病変が髄膜表面から脊髄の内側へ広がっていく傾向があるため中心灰白質よりも白質の症候が出やすいと考えられている。脊髄症で発症するサルコイドーシスは下記の3パターンが多い。
- MRIで脊髄腫大を認める亜急性進行性脊髄症
この場合は視神経脊髄炎との鑑別が重要になる。症状のピークに達するのに4週間以上かかる場合は視神経脊髄炎ではなく腫瘍やサルコイドーシスが想定される。また脊髄サルコイドーシスが下部胸椎レベルに認められるときはdural AVFが鑑別にあがる。
- 頚椎症による脊髄圧迫があるが、髄内高信号が広範囲に認められる場合
脊柱菅狭窄症で脊髄は腫大しないため脊髄の腫大が認められる場合は脊髄サルコイドーシスの可能性も考える。また病変部に一致した背部痛はサルコイドーシスに多く認められ、およそ6割程度といわれている。
- 脊髄腫瘍に類似した髄内結節を認めるとき
末梢神経サルコイドーシス
サルコイドーシスの末梢神経障害で最も多いのが視神経や顔面神経などの脳神経の障害である。脳神経麻痺は神経サルコイドーシスの75%を占める。脳神経麻痺を除いた末梢神経の障害、すなわちサルコイドニューロパチーは神経サルコイドーシスの15%程度と言われているが様々な報告がある[79]。サルコイドーシスの中で致死的な転帰をとるのが1~5%であり、その原因として多いのは肺と神経の障害である。神経障害の中で致死的になりやすい因子として末梢神経障害が挙げられており[80]、サルコイドニューロパチーは治療の標的として重要である。
サルコイドニューロパチーの病理像では神経上膜を中心に肉芽腫と壊死性血管炎の所見に加え、血管炎ニューロパチーで見られるような、急性軸索変性像と神経束ごとの有髄神経線維密度の偏りが報告されている[10]。このことからニューロパチーを生じる機序としてサルコイド結節による神経線維への圧迫や、神経線維に血液を供給するvasa nervorumに生じた血管炎機序に伴う梗塞が推測されている[81][82]。一方、サルコイドニューロパチーでは非常に稀ながら神経伝導検査で脱髄性ニューロパチーを呈することがある。その機序としては何らかのサイトカイン・免疫因子の関与や[82]神経内鞘内に存在するサルコイド肉芽腫よって圧迫され伝導ブロックを生じた可能性が想定されている[83]。
サルコイドニューロパチーには2つの主要なパターンがある[15]。その2つとは四肢遠位部優位の感覚運動型ポリニューロパチー型と四肢近位部と遠位部とが同等に障害されるnon-length dependentの左右非対称性多発神経根ニューロパチー型である。多発性単神経障害と診断される例は少ない[82][84]。左右非対称性多発神経根ニューロパチー型では髄液所見や神経根のMRI所見に異常が認められることがある。発症経過としては急性から亜急性の経過をとることが多く、数週から数ヶ月かけてプラトーに達するのが典型的であり自然軽快傾向を示すことも珍しくない[81][84]。疼痛やジンジン感など陽性の感覚障害が目立つことが多く、逆にこれらの症候がない場合はには他の原因の可能性が高い[84]。陽性の感覚障害は小径線維ニューロパチーないし血管炎ニューロパチーとしての特徴と考えられている。non-length dependentの左右非対称性多発神経根ニューロパチー型の場合はギラン・バレー症候群やCIDPとの鑑別が重要となる。
急性の経過でギラン・バレー症候群と類似した臨床像を呈したサルコイドニューロパチーが複数報告されている[10][82][84][85][86][87]。ギラン・バレー症候群との鑑別に有効な所見としてサルコイドニューロパチーでは、神経症候が左右非対称性、多臓器障害を反映した全身症状を伴いやすい、脳脊髄液での細胞数増多、免疫グロブリン大量療法が無効ないし効果不十分、ステロイドが有効、回復期に再燃などが重要と考えられる。
少数であるが脱髄所見が主体のサルコイドニューロパチーはCIDPとの鑑別が重要になる。神経伝導検査で伝導ブロック様の波形変化を示し脱髄性障害が示唆され、CIDP類似の臨床像を示したサルコイドニューロパチーが9例報告されている[10][83][88][89][90][91]。伝導ブロック様の波形とは遠位刺激のCMAP振幅は低下しているが近位刺激のCMAPは振幅が保たれ、時間的分散が目立たない状態である。これらの症例では四肢筋力低下が左右非対称や多発単神経障害型を示す、脳神経麻痺を伴う、サルコイドーシスによる多臓器障害を伴う、副腎皮質ホルモンが有効といった特徴があった。MADSAMが重要な鑑別疾患になる。CIDP疑いの症例で免疫グロブリン大量療法に抵抗性の場合、その原因が免疫治療が不十分である可能性に加え[92]、サルコイドニューロパチーを含めた他の疾患の可能性を想定するべきである。特に免疫グロブリン大量療法や血液浄化療法が無効でステロイドが著効する場合にはサルコイドニューロパチーを鑑別の上位に置く必要がある[91]。
筋サルコイドーシス
筋サルコイドーシスはサルコイドーシスの肉芽腫病変が全身の骨格筋組織に出現した場合に診断される[93][94]。筋サルコイドーシスは無症候性と症候性に大別される。多くが無症候性であり、症候性は稀な病態でありサルコイドミオパチーと呼ばれる。サルコイドミオパチーは腫瘤型とミオパチー型に分類され、さらにミオパチー型は発症様式から急性筋炎型と慢性ミオパチー型に分類される[93][94][95]。慢性ミオパチー型のうち孤発性封入体筋炎を合併する特殊なサブグループの報告が散見する[96][97][98]。全身性サルコイドーシスの患者における無作為の筋生検で50~80%類上皮細胞肉芽腫を認めたと報告され、無症候性であっても筋病変を有すると考えられている[99]。
症候性のサルコイドミオパチーは全身性サルコイドーシスの0.4~2.3%と報告され極めて少ない[93][100][101]。サルコイドミオパチーは日本では腫瘤型73%、ミオパチー型27%であり腫瘤型が多い[93]。筋組織に限局したサルコイドーシスも報告されている[102]。診断には筋生検が重要である[44]。
病型
- 無症候性
無症候性は腫瘤の触知や筋痛、筋力低下、筋萎縮など自覚的にも他覚的にも筋症状はなく、筋生検や画像検査で偶然発見されるものを示す。
- 腫瘤型
腫瘤型は筋腫瘤の触知で気が付かれる。自発痛や圧痛、把握痛が半数に認められる。多くは筋力低下や筋萎縮を示さないが腫瘤により神経圧迫し筋力低下や筋萎縮を示すことがある。
- ミオパチー型
急性筋炎型と慢性ミオパチー型に分けられるが発症様式の相違を除けば、この2つは類似している。ミオパチー型では筋腫瘤はふれず、腫瘤型の経過中にミオパチー型に移行することはないと言われている。どちらも近位筋優位に筋力低下、筋萎縮、自発痛、把握痛が認められる。臨床所見から他の炎症性筋疾患と鑑別は困難であるが筋生検では類上皮細胞肉芽腫が認められるため区別可能である。
- 孤発性封入体筋炎合併例
孤発性封入体筋炎の臨床的、病理学的特徴を有するサルコイドミオパチーが報告されている。深指屈筋など遠位筋の筋力低下が認められ、細胞質5’-ヌクレオチダーゼ(cN1A)に対する自己抗体が一部の症例で検出された。筋生検では肉芽腫病変に加えて、炎症細胞浸潤、筋線維の大小不同、線維化などサルコイドミオパチーの所見に加えて、縁取り空胞やアミロイド沈着、コンゴーレッド陽性物質、HLA-ABC陽性筋線維など孤発性封入体筋炎の病理学的特徴も認められる。通常のサルコイドーシスよりもステロイド抵抗性の症例が多いとされている。
治療
腫瘤型は経過観察して良いという意見もある。腫瘤型筋サルコイドーシスは、プレドニゾロンを30~40 mg開始し徐々に漸減で軽快するが、プレドニゾロンの減量や中止によって再燃する例も知られている。どの時点で治療を開始し、中止するのか明確になっていない。
心臓サルコイドーシス
サルコイドーシス患者における心臓病変の頻度は、5~10%程度とされている。サルコイドーシス剖検例の20~27%に心サルコイドーシスが認められ、生前の診断率は40~50%程度である。心サルコイドーシスと診断されていないサルコイドーシスの患者も、精査をすると40~50%程度に心サルコイドーシスが見れられるとも言われたり、高齢の日本人女性の場合は80%程度が心サルコイドーシスを合併していると言われたりするなど、その合併頻度に関してはバラつきが多い。
心サルコイドーシス診断には、心電図、ホルター心電図、心シンチグラフィィ、MRI、心臓カテーテル、心筋生検、PETなどを用いるが、示す病像が病期や重症度に応じて多岐に及ぶため診断は容易ではない。例えば、心筋生検を行うと、病変部が採取できていれば、乾酪壊死を伴わない類上皮細胞肉芽腫を始め、ラングハンス型巨細胞(星芒小体やSchaumann小体を持つ)、異物型巨細胞やリンパ球浸潤が認められる。しかし、サルコイドーシスは結節性の疾患であり、心筋内に散在性に病変が認められるため、心筋生検を行っても、心サルコイドーシスの病変部が含まれない可能性もある。また、心筋生検を行った事が不整脈を誘発し得るので、注意を要する。採取したサルコイドーシスの心筋を電子顕微鏡で観察すると、心筋内毛細血管の基底膜の多層化が認められ、ミクロアンギオパチーの機序による病態も提唱されているが、この所見は糖尿病でも認められサルコイドーシスに特異的ではない。
心サルコイドーシスは、重症心不全や致死的不整脈を引き起こし得て、サルコイドーシスによる死因として上位の合併症である。心サルコイドーシスによる不整脈は、脚ブロックや房室ブロックから洞不全症候群などの致死性不整脈まで進行する場合もある。
サルコイドーシス関連心不全を呈する患者の心臓は、拡張型心筋症(DCM)を呈するのが一般的である。ただし、日本では特発性DCMの5年生存率が64%である一方で、心サルコイドーシスの5年生存率は37%と低い。2006年に改訂されたサルコイドーシスの診断基準と診断手引では、心臓サルコイドーシスに比較的特徴的である完全房室ブロック、心室中隔基部の菲薄化、心臓へのガリウムの異常集積、左室収縮不全が主徴候とされ、新たに造影MRIの遅延増強所見が加えられた。遅延増強効果は、活動性炎症部位の評価や、ステロイド系抗炎症薬投与による治療効果の判定にも有効である。心サルコイドーシスのステロイド系抗炎症薬の全身投与の適応は、高度房室ブロック、心室頻拍などの重症心室不整脈、局所壁異常運動、あるいはポンプ失調とされている。その治療効果は房室ブロックでは伝導障害が改善し正常化する例が有り、低心機能例では収縮能は改善しないまでもそれ以上に悪化しない例が多い。また低収縮に至る前に治療を行った場合は、改善する例も知られている。ステロイドの投与量はPSL30mg/dayまたは60mg/2dayが推奨されている。4週間投与したのち、2~4週間毎に漸減していくことが多い。ステロイドの中止に関しては明確な規定は存在しないが最終的にPSL10mg/day程度での維持療法を行う場合が多い。
なお、重症不整脈に関してはペースメーカー、カテーテルアブレーション、植え込み型除細動器などが用いられる。
皮膚サルコイド
サルコイドーシス患者におけるサルコイドーシスの皮膚病変の発生頻度は、10~30%程度である。これは、胸郭内病変、リンパ節病変、眼病変に次ぐ。サルコイドーシスの発見契機となる自覚症状としては、眼病変に次いで2番目に多い。皮膚の場合は、他の臓器と比べて生検が行い易く、生検を行って診断を行う。日本では福代の分類に従って記載される。福代の分類は、組織学的特徴を加味した分類であり、結節性紅斑、瘢痕浸潤、皮膚サルコイドに大別される。皮膚サルコイドはさらに結節型、局面型、びまん浸潤型、皮下型、その他の稀な病型に分類される。皮膚サルコイドの中では結節型が最も多く、局面型、皮下型がそれに次ぐ。結節型、局面型ともに顔面に好発し、皮下型は四肢に多発する。組織像は類上皮細胞が種々の大きさの肉芽腫を形成する。肉芽腫の大きさの分布は、病型によって異なる。皮膚病変の治療の原則は、ステロイド系抗炎症薬の外用である。ただし、strong以下の外用薬は著効しない。なお、ミノサイクリンの長期投与で皮膚サルコイドーシスが寛解したという報告が有る。
- 結節性紅斑
- 結節性紅斑は、肉芽腫を認めない、発赤を伴った有痛性の皮下硬結が、両側下腿伸側に多発した病変である。初期には好中球浸潤、後期には多核巨細胞が認められることがあるものの、肉芽腫は認められない。
- ただし、これは非特異的病変であり、これが見られてもサルコイドーシスとは限らない。
- 瘢痕浸潤
- 肉芽腫と共に異物が証明される病変である。陳旧性の瘢痕に、肉芽腫反応が認められる。紅褐色の丘疹、結節、あるいはそれらが癒合した病変である場合が多い。外傷を受け易い、膝蓋、肘、顔面に好発する。組織学的には類上皮細胞性肉芽腫に加えて、偏光顕微鏡で病変部に重屈折性を示す異物が認められる。
- 皮膚サルコイド
- 肉芽腫を認め、サルコイドーシスに特異的な病変である。結節型、局面型、びまん浸潤型、皮下型、その他の稀な病型に分類される。
- 結節型
- 隆起型の病変であり、皮膚サルコイドの中で最も頻度が高い。紅色の丘疹、結節で、鱗屑や血管拡張を伴う場合が多い。顔面、四肢に好発する。なお、顔面では鼻周囲に生じる例が多い。組織像では真皮全層に大型の結節が認められる。
- 局面型
- 水平方向に伸展する病型であり、環状の形態を示す場合と斑状の病変とがある。なお、環状病変が多い。
- 環状病変は辺縁がやや堤防上に隆起し、中央部は正常皮膚色でやや萎縮性である。種々の大きさを示し、場合によっては、手掌大に達する例もある。なお、前額部に好発し、多発する傾向がある。組織像は真皮上層に比較的小さな肉芽腫が認められる。
- なお、局面型の皮膚病変が見られる場合には、心サルコイドーシスの合併率が、他の皮膚病変と比べて高い。
- びまん浸潤型
- lupus pernioに相当する病変である。組織像は、小型の肉芽腫が真皮全層に散在し、血管拡張を伴う。自然寛解する例は稀で、遷延する場合が多い。
- 皮下型
- 皮下の弾性硬の結節、硬結であり、表面の皮膚は通常の皮膚色と変わらない。多発傾向で、四肢に好発する。組織像は皮下脂肪組織中に局在する。稀に、顕著な壊死が認められる。
その他の臓器障害
肝病変
サルコイドーシス患者に腹腔鏡・肝生検を実施すると、80%の高頻度で潜在性の肉芽腫性病変が認められる。腹部CTでは、転移性肝悪性腫瘍を疑わせる、多発性結節性低吸収域として認められる。ただし、生化学所見や臨床所見で肝病変の証拠が有っても、生検を行う事は稀である。
脾病変
潜在性の脾病変は、サルコイドーシス患者の38~77%で認められる。CTでは多発性低吸収域や形態異常を示す。
高カルシウム血症
サルコイドーシスに高カルシウム血症が高頻度に合併する事は、古くから知られている。サルコイドーシスや結核では、肺胞マクロファージや肉芽腫の類上皮細胞がビタミンD-1α水酸化酵素を産出し、1,25(OH)2Dが過剰に変換されるために、消化管でのカルシウムの吸収率が増加し、また骨からのカルシウムの動員が行われ易くなり、高カルシウム血症が起こる。
サルコイドーシスに伴う肉芽腫の類上皮細胞でのビタミンD-1α水酸化酵素の増加の場合は、副甲状腺ホルモン(PTH)による支配は受けずに、IFNγによって産出は亢進され、ステロイドホルモンによって抑制される。すなわち、ステロイド系抗炎症薬の投与によって、サルコイドーシスに伴う高カルシウム血症は改善する。つまりサルコイド肉芽腫自体が、内分泌器官として働いているとも言える。PTHrpとの関連は不明な点が多い。
腎病変
サルコイドーシスでは、高カルシウム血症による腎障害、尿細管間質性腎炎、肉芽腫性間質性腎炎、糸球体腎炎、腎血管炎が稀に起こる。
内分泌病変
間脳-脳下垂体系は神経系との接点であり、中枢神経病変としてもサルコイド肉芽腫がよく見られる部位である。血管壁や血管周囲に肉芽腫が形成される。
出典
参考文献
関連項目
外部リンク

